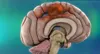
What Is Glioblastoma?
Glioblastoma is an aggressive cancer that usually starts in the brain. Learn how this fast-growing brain tumor spreads.
You can also search by physician, practice, or hospital name
Find your WebMD Facebook community to learn, share and connect.
Use this guided discussion to help you navigate chronic myeloid leukemia (CML) treatment options, ask informed questions, and collaborate with your healthcare provider for personalized care.
Join Group