1 of 4 / Teeth and Gums
View All
Smoking and Oral Health
Smoking and other tobacco use can cause oral health problems like gum disease and tooth decay.
Tooth Decay Prevention
Tooth decay is the destruction of tooth structure and can affect both the enamel and the dentin layer of the tooth.
Toothaches
Toothaches and jaw pain are common but can have different sources. Learn more from the experts at WebMD.
Sensitive Teeth
Tooth sensitivity is discomfort in one or more teeth that is triggered by hot, cold, sweet or sour foods, or drinks, or cold air.
2 of 4 / Other Oral Conditions
View All
Changing Bad Breath
Bad breath is often caused by bacteria in your mouth that gives off noxious odors or gases that smell like sulfur.
Bad Breath (Halitosis)
Bad breath, medically called halitosis, can result from poor dental health habits and may be a sign of other health problems.
Dry Mouth Overview
We need saliva to moisten and cleanse our mouths and digest food. When we don't produce enough saliva, our mouth gets dry.
Dry Mouth and Other Side Effects of Medications
All drugs, whether taken by mouth or injected, come with a risk of side effects. Learn which meds might cause oral side effects.
3 of 4 / Dental Care Basics
View All
Dentists and Other Oral Health Care Providers
Many different types of oral health care providers could become involved in the care of your teeth, gums, and mouth.
Teeth and Gum Care
The healthier your teeth and gums are, the less risk you have for tooth decay and gum disease.
Finding a Dentist
You and your dentist will be long-term oral health care partners; therefore, you should find someone you can be comfortable with.
Oral Piercings: Tongue, Lip, and Cheek
While piercing the tongue, lip, or cheek may be attractive to some, there are many health risks associated with oral piercing.
4 of 4 / Treatment & Surgery
View All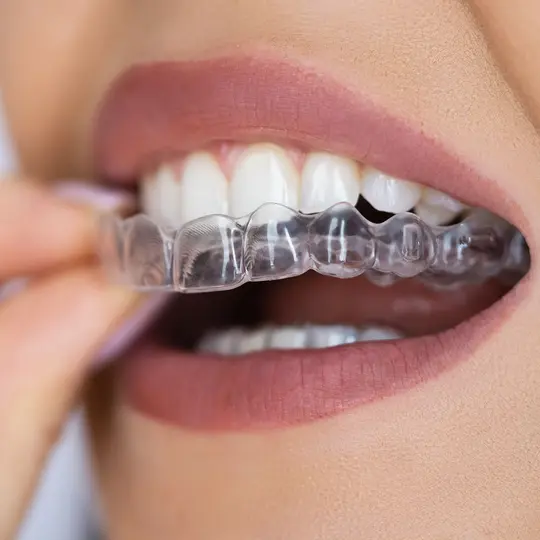
Crooked Teeth and Misaligned Bites
Most often crooked teeth, overbites, and underbites are inherited traits just as the color of your eyes or size of your hands.
Invisible Aligners for Teeth
More and more people are having success with clear orthodontic devices called aligners.
Braces and Retainers
If you have crooked teeth and/or a misaligned bite, braces and retainers can help straighten teeth.
Oral Surgery
Find out what conditions might require oral surgery and what to expect.
Suggested Reads about Oral Health
Toothbrushing Lowers Pneumonia Risk in Hospital: Study
Sometimes, a hospital stay results in developing a new health problem. About 1 in 100 people develop pneumonia while in the hospital, but a new study reports that a daily tooth-brushing regimen during hospital stays significantly reduced the likelihood of developing the potentially life-threatening infection.
Tissue Regeneration Showing Promise as Alternative to Root Canal
Scientists are developing a new dental treatment – tissue regeneration -- that could replace the root canal, according to a statement from the ADA Forsyth Institute.
The 5 Things Dentists Wished Doctors Weren’t Missing
Dentists are urging primary care doctors to pay closer attention to signs of illness that may show up in the mouth. From overlooked gum disease to suspicious lesions, oral health can provide a critical window into broader medical concerns.
Lead Aprons No Longer Needed During X-Rays, Dental Group Says
Wearing a heavy lead apron is no longer necessary during routine dental X-rays, a professional dental radiology group has concluded.
Top Search Terms for Oral Care
8 million+ Physician Ratings & Reviews
Find Doctors and Dentists Near You
You can also search by physician, practice, or hospital name